- 移动端


北京擎科生物科技股份有限公司品牌商
7 年
手机商铺
- NaN
- 0.5
- 0.5
- 1.5
- 0.5

北京擎科生物科技股份有限公司
入驻年限:7 年
- 联系人:
张楠
- 所在地区:
北京 大兴区
- 业务范围:
实验室仪器 / 设备、试剂、技术服务、细胞库 / 细胞培养、抗体、ELISA 试剂盒、耗材
- 经营模式:
生产厂商 科研机构





公司新闻/正文
干货|RNA-Seq应用案例分析
3651 人阅读发布时间:2021-10-15 11:38

当某一组织或细胞处于异常环境下,或者由于自身变异或者受到外部环境的刺激,如极端环境(高温、低温)、细菌病毒感染等,这往往会导致RNA的表达发生变化,从而影响之后蛋白表达的变化,导致一系列的病理反应的产生。而RNA-Seq技术能够在单核苷酸水平对特定物种的整体转录活动进行检测,从而全面快速地获得该物种在某一状态下的几乎所有转录本信息。这就是RNA-Seq分析的意义。
通过对转录组数据分析,可以对以下信息进行挖掘:
-
转录本结构研究:检测未知转录本和稀有转录本、转录本结构研究。
-
非编码区域功能研究:如microRNA、非编码长RNA (lncRNA)、RNA编辑。
-
基因转录水平研究:如样本基因表达量、实验中对照组间差异表达。
-
转录本结构变异研究:如可变剪接、融合基因鉴定、编码序列多态性研究。
-
开发SNPs和SSR等。
RNA-Seq可以快速获得目的细胞、组织或生物体内的mRNA种类以及其丰度,分析结果可以有助于发现由于可变剪接等所产生的新转录本;快速、高效获得所研究的不同细胞或者不同组织内的mRNA种类及其丰度,分析mRNA的差异表达信息。通过差异表达基因功能注释信息,得到相关功能通路结果,从而找到与研究相关基因,进行后续验证。如果想要研究某种基因如何通过改变细胞的基因表达调控网络来发挥其生物学功能,可以对该基因进行突变、敲除或敲低,在对照组和实验组的细胞内进行RNA-seq分析,通过差异表达分析即可以快速全面获得需要的信息。
案例1:哺乳动物组织转录组分析
哺乳动物由于其基因组大小很大并且很复杂(高重复、高杂合),导致其转录组研究比较困难。在“Mapping and quantifying mammalian transcriptomes by RNA-seq”中Mortazavi等研究员结合Illumina测序平台,对成年小鼠的脑、肺、骨骼肌组织RNA进行转录组高通量测序及分析(4100-5200万个25碱基对图谱),大于90%的位置与已知外显子相匹配,同时也发现了新的和未修改过的基因,包括改变或增加启动子、外显子和3’未转录区域。约3000个新鉴定的3’UTR,可能在microRNA介导的转录后水平和翻译水平调控中起重要作用;约3000个新鉴定的5’外显子,提示有新的启动子序列被利用。尤其在RNA剪接方面,该研究利用高通量测序获得的海量数据,对比到约2×10^5种可能剪接方式的数据库,鉴定出1.45×10^5种不同的剪接方式,其中可变剪接占主导,3500个基因拥有至少一种内部剪接方式。
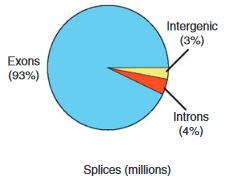
图1.Reads比对区域占比
该研究成果表明,RNA-Seq技术不仅能够检测到低丰度转录本,而且可以发现未知转录本,精确识别可变剪接位点,提供全面的转录组信息,是目前深入研究转录组复杂性的有力工具。
案例2:物种间表达差异
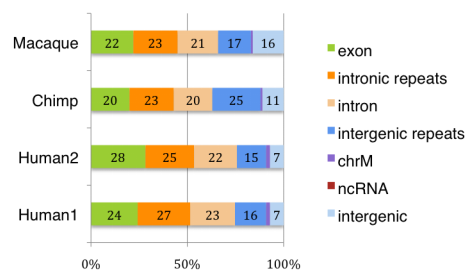
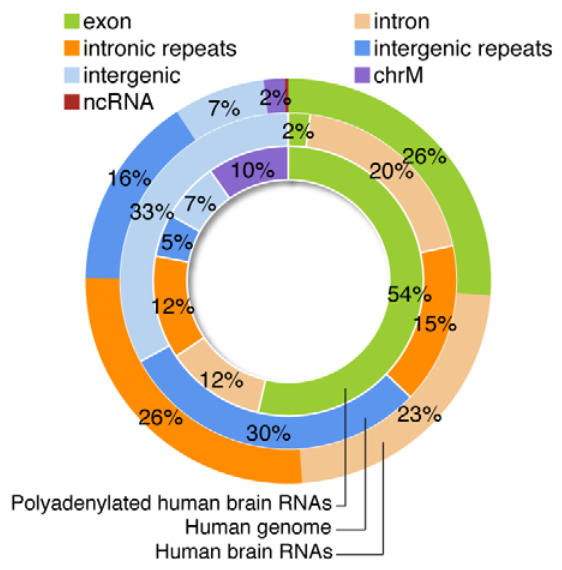
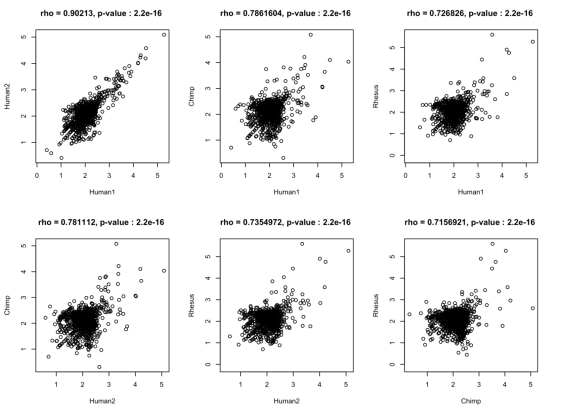
虽然人类大脑中注释基因的表达已经被广泛地描述,但我们对位于已知基因边界之外的转录本的范围和保存的知识是有限的。在“Intergenic and Repeat Transcription inHuman, Chimpanzee and Macaque Brains Measured by RNA-Seq”中Augix Guohua Xu等研究者利用RNA-Seq技术研究了人、黑猩猩和恒河猴三个物种的大脑在不同年龄段的基因表达量。所有物种中,只有20–28%的转录本可以比对到注释的外显子区,20–23%的转录本比对到内含子上,来自内含子和基因间区的重复序列占大脑转录组的40–48%。一些重复家族表现出转录本拷贝数升高。在非重复基因间区,研究者鉴定出1093个在人大脑中显著高表达的区域。这些区域在灵长类RNA表达水平和哺乳动物DNA序列水平都具有保守性。20%的转录本在已知基因的3’UTR有延伸,这可能在可变microRNA调控基因表达中发挥作用。最后,研究者发现物种间的转录组表达差异随着进化时间的增加而逐渐增大,相比外显子,基因间区转录本表现出更大的差异表达。
该研究结果显示,RNA-Seq技术发现人大脑中存在很多尚未鉴定出的进化保守的转录本,并且其中一些转录本可能在转录水平调节和人特有的表型特征进化中起作用,为从分子水平上揭示大脑发育机制提供了有效方法。
以下为部分结果展示:
左右滑动查看更多图表
案例3:水稻转录组分析
在真核生物中,掌握转录组的动态变化对研究转录调控的复杂性以及转录调控对表型的影响至关重要。在过去的几十年里,水稻因其农业价值而被广泛研究。在“Deep RNA sequencing at single base-pair resolution reveals high complexity of the ricetranscriptome”中Zhang等研究者运用RNA高通量双向深度测序技术,首次展现了水稻八个组织部位的转录本表达图谱全貌,并且能够精确检测到丰度非常低的转录产物,获得相当可观数量的全新转录本、外显子、非编码区。经生物信息学基础分析和深度分析,揭示了顺式剪接在水稻基因中占33%以上,234个推测嵌合转录本可能由反式剪接产生,大量融合转录本可能是可变剪接的副产品,其数据量和信息量远远高于已有报道。RNA-Seq技术使水稻转录组信息得到了极大的丰富,帮助研究者更全面的认识转录本的多样性和复杂性,拓展了未来农业研究的领域。
案例:病毒侵染过程中细胞基因表达模式的改变
病毒侵染过程中除了病毒基因的激活,通常还伴随着细胞基因表达的紊乱。在“Simultaneoushigh-resolution analysis of vaccinia virus and host cell transcriptomes by deepRNA sequencing”中Zhilong Yang等研究者利用RNA深度测序技术对感染牛痘病毒(VACV)后不同时段的VACA病毒和HeLa细胞的转录组进行了同步研究。测序共得到大约5亿条短cDNA序列,构建了不同感染时间的完整VACV转录组和超过14,000条的宿主mRNA。病毒DNA复制前,检测到118个开放阅读框(ORFs)的转录本;复制后,又检测到另外93个开放阅读框的转录本。高通量测序技术使得很多mRNA的边界得以精确的界定。按感染时间分析,与DNA合成一样,在蛋白质抑制物存在时合成早期mRNA的两个簇,暗示他们和其他DNA病毒不一样,不分即早期和晚早期发展阶段。在4h时,病毒编码的mRNA占总转录RNA的25–55%。这一快速的变化,导致大量宿主mRNA含量下降,进而导致宿主蛋白质合成的急剧下降而丧失抗病毒能力。但是在2h时,宿主mRNA有小幅度的增加,这些上调RNA为NF-κBcascade,凋亡、信号转导、配体介导的信号因子,似乎参与宿主对病毒浸染的应答反应。
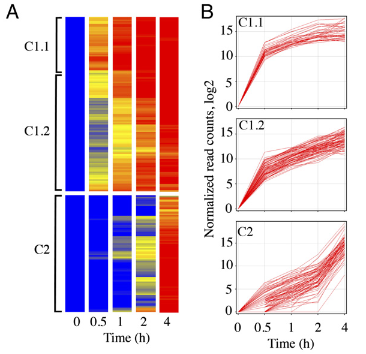
图2.药物处理前4小时表达热图及对应折线图
如有高通量测序需求,回复小编或联系ngs@tsingke.com.cn即刻开聊,下单可直接联系当地销售经理。